Notice
UW Health recently identified and investigated a security incident regarding select patients’ information.Learn more
UW Health recently identified and investigated a security incident regarding select patients’ information.Learn more



Overview
When you are living with cystic fibrosis, you need access to daily support and regular care. The UW Health Kids team at the UW Health Cystic Fibrosis Center provides nationally recognized treatment for children and adults.
You receive services from doctors with specialty training in lung disease. We are one of 120 centers in the U.S. accredited by the Cystic Fibrosis Foundation.
Our cystic fibrosis care teams teach other health care professionals about cystic fibrosis and take part in research to provide better diagnosis and treatments.
Programs and research
When you seek care from the UW Health Kids team at the UW Health Cystic Fibrosis Center, we work with you to:
Educate your family about providing the care and help you need
Promote your good health
Prevent your condition from getting worse
Help you live the life you want
The National Institutes of Health recognizes our center as a model of effective and efficient health care for a chronic disease.
Our patients enjoy healthy outcomes for lung function that are higher than the national average.
The cystic fibrosis doctors and scientists at UW Health lead and participate in many research projects and clinical trials. We want to provide better treatments and improve the care you receive.
The UW Health Cystic Fibrosis Center is one of more than 115 care centers across the country accredited by the Cystic Fibrosis Foundation. We work closely with the Foundation to continue to improve the care and quality-of-life of our patients and publicly report our center-specific data to give patients, families and ourselves an opportunity to look for areas of improvement.
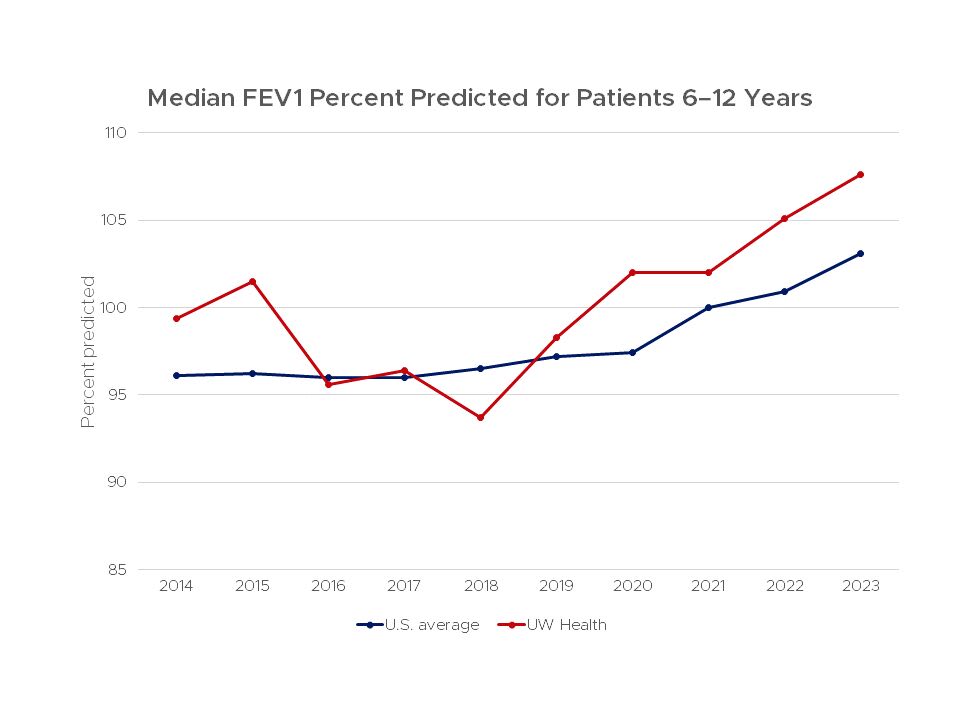
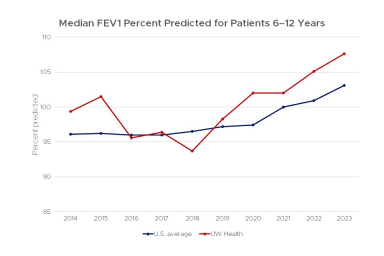
Pulmonary function outcomes are important indicators of the health of cystic fibrosis patients. The forced expiratory volume in 1 second (FEV1), the volume of air a person can forcefully blow out in a second is considered a good indicator of lung function.
This chart shows lung function for patients age 6–12 that were treated at the UW Health Cystic Fibrosis Center compared to the average score for all centers in the United States from 2011–2022
.
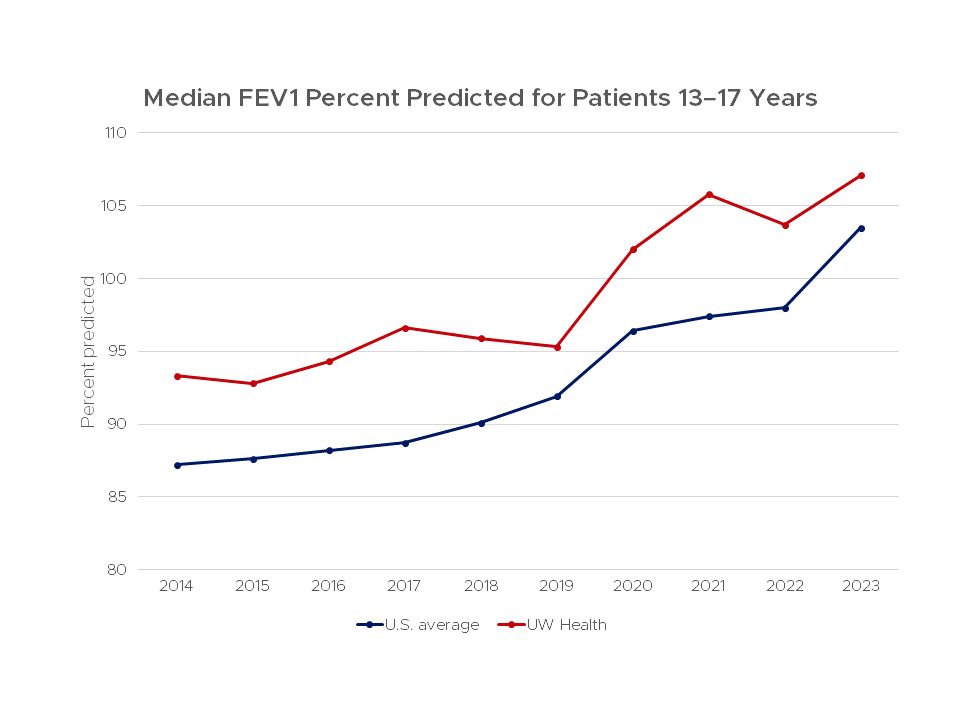
Pulmonary function outcomes are important indicators of the health of cystic fibrosis patients. The forced expiratory volume in 1 second (FEV1), the volume of air a person can forcefully blow out in a second is considered a good indicator of lung function.
This chart shows lung function for patients age 13-17 that were treated at the UW Health Cystic Fibrosis Center compared to the average score for all centers in the United States from 2011–2022.
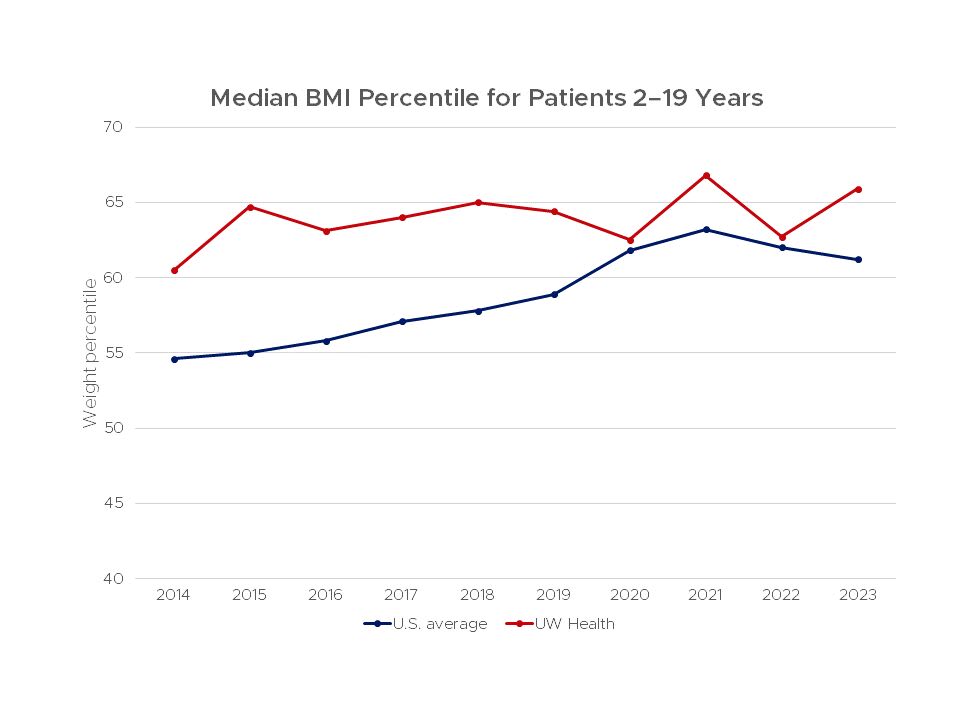
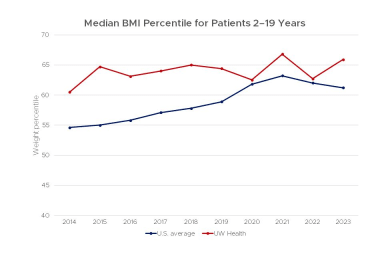
The Body Mass Index (BMI, a ratio of body weight to stature) is considered a good indicator of nutritional status.
This chart shows BMI for patients treated at the UW Health Cystic Fibrosis Center compared to the average score for all centers in the United States from 2011–2022.
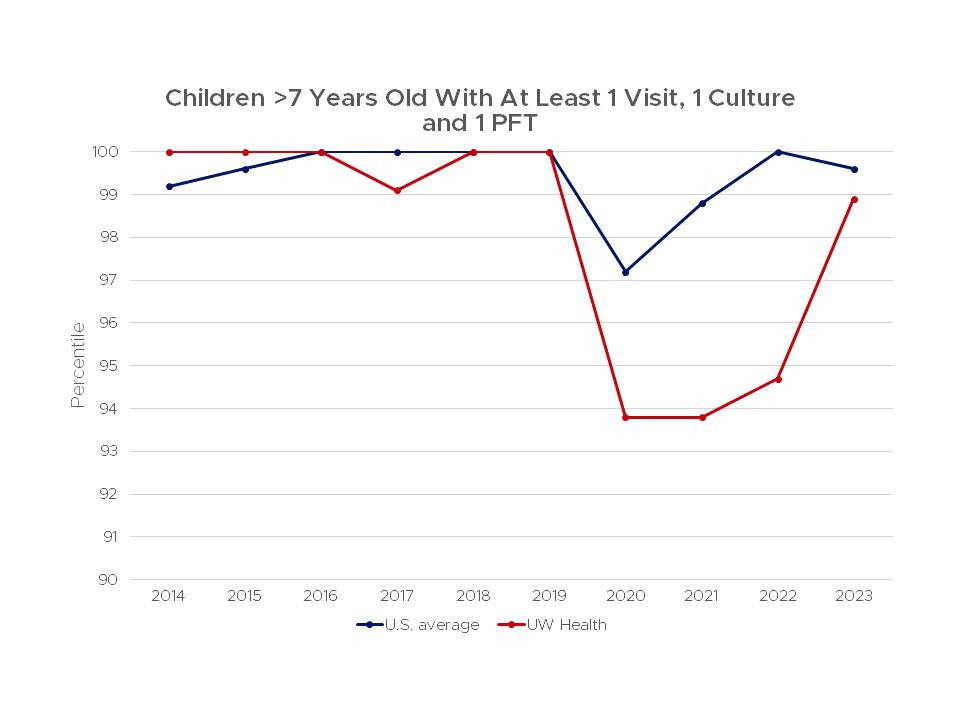
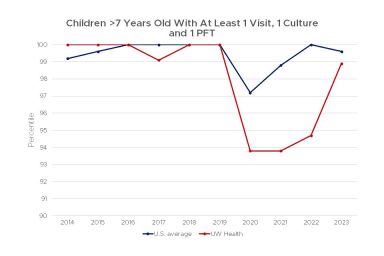
Current guidelines of care recommend that patients with cystic fibrosis be evaluated at an accredited CF Center at least quarterly. In addition, patients should undergo pulmonary function testing at least two times a year and have a culture of their respiratory secretions performed at least once a year. The recommendation is also to increase the frequency of follow up for those patients that have more severe disease.
We believe that more frequent clinic visits and more frequent monitoring of pulmonary function and sputum cultures can lead to better outcomes. Thus, we strive to exceed the Cystic Fibrosis Foundation's recommended testing and obtain pulmonary function test and a sputum culture (or throat swab culture for patients who cannot cough out sputum) at every clinic visit.
This chart shows the percent of children over 7 years old who had at least 1 visit, 1 culture and 1 PFT at the UW Health Cystic Fibrosis Center, compared to the average for all centers in the United States from 2011–2022.
Conditions and symptoms
The UW Health Kids team at UW Health diagnoses patients with cystic fibrosis at birth and in adulthood. Most people with cystic fibrosis know they have the condition as children.
More adults with less common symptoms are being diagnosed later in life. You might live with chronic coughing and sinus infections and not be able to gain weight.
We use a simple sweat test to make a diagnosis. We place two electrodes on your forearm. This causes your sweat glands to make sweat. We check your sweat for high levels of chloride. Your doctor may order additional tests such as blood work and X-rays to confirm the diagnosis.
If you have cystic fibrosis, your body makes thick, sticky mucus. This mucus can affect how your lungs, pancreas, sweat glands and reproductive system work. Cystic fibrosis is a genetic condition. Symptoms are different for everyone.
Chronic or persistent cough
Frequent lung infections
Poor weight gain
Frequent loose stools
Frequent lung infections
Salty-tasting sweat
Arthritis
Infertility
Pancreatitis
Sinusitis
You might experience complications related to your cystic fibrosis as you get older. These conditions can include:
Cystic fibrosis-related diabetes
Intestinal blockage
Liver disease
Nasal polyps
Osteoporosis
Treatments
You need daily treatments and regular care to stay healthy and live well.
Daily treatments include:
Airway clearance therapies to loosen mucus and clear it from your airways, such as:
Active cycle breathing
Autogenic drainage
Forced expiration technique
Manual chest physiotherapy
Exercise
Nutrition therapy, including enzymes and vitamin supplements
Other available treatments include:
Fertility treatments
For some individuals with advanced disease, lung transplantation might be the next step to help regain health.
You can find a balance between your busy lifestyle and your care. The team at UW Health Kids knows that living with cystic fibrosis can be emotionally and physically challenging. Your care team stays by your side to help you feel your best.
Some tips for staying healthy with cystic fibrosis:
Avoid others with upper respiratory illness
Complete your physiotherapy exercises
Contact the cystic fibrosis clinic if there are changes in your health
Eat a cystic fibrosis-specific diet high in calories and fat
Keep yourself hydrated
Manage your mental health
Stay active
Take your prescribed medication
Talk to your care team about annual flu and pneumonia vaccines
Visit the cystic fibrosis center regularly throughout the year
Wear a mask that covers your nose and mouth when in a health care setting

Patient resources
Watch videos from Cystic Fibrosis Family Education Day.
If a newborn screen test shows your baby might have cystic fibrosis (CF), a sweat test will make the final determination.
For patients having telemedicine/video appointments, we offer throat swab testing at a drive-through site.
For patients over 6 years of age who are having telemedicine/video appointments, learn how to set up and use a spirometer.
Parents can take several steps to keep their infants and young children with cystic fibrosis healthy:
Parents who smoke are encouraged to quit or at least not smoke in the home or car.
Each fall a flu shot is recommended for the person with cystic fibrosis and other family members.
During hot weather or times of physical exertion, parents should add salt to the child's food.
Regular aerobic exercise is encouraged to increase the child's oxygen intake, strengthen lung muscles and increase their sense of well-being.
Parents are also encouraged to look beyond the disease to see their children as unique individuals who happen to have cystic fibrosis. Children should be encouraged to pursue their interests and personal goals. Children with cystic fibrosis participate in sports, dance, music, outdoor activities or scouts, as well as their regular school activities.
Printable passport for cystic fibrosis patients to inform care providers about their diagnosis and precautions that should be taken while treating them.
Learn more about our current research studies
Meet our team
The UW Health Kids pulmonology team includes experts in lung disease and cystic fibrosis nutrition.
Locations
Provider resources
In Wisconsin, cystic fibrosis (CF) is one of 47 disorders for which infants are routinely screened by a heel prick during the first few days of life.
An abnormal cystic fibrosis screening test does not mean a child has cystic fibrosis. It means the child might have cystic fibrosis. An additional test, called a sweat test, is needed to determine whether the child has cystic fibrosis.
The newborn-screening test for cystic fibrosis involves two steps. First, blood obtained through routine newborn screening is examined for trypsinogen, a substance found to be higher in infants with cystic fibrosis. If the trypsinogen level is elevated, a second test is done on the blood sample to examine it for cystic fibrosis gene mutations.
There are several ways in which the cystic fibrosis screening test can be abnormal:
If the trypsinogen level is extremely elevated but there is no cystic fibrosis gene mutation, there is a small possibility (less than one in 100) that the child has cystic fibrosis. Although the newborn screening test looks for 23 different cystic fibrosis gene mutations, other cystic fibrosis gene mutations exist that are not identified through newborn screening. A small possibility exists that the child could have two of those other cystic fibrosis gene mutations. Some possible reasons (other than the presence of cystic fibrosis) for elevated trypsinogen levels include a stressful or premature delivery and/or low Apgar scores. If the child has signs of cystic fibrosis or a family history of cystic fibrosis, then the sweat test is needed to be sure the child does not have cystic fibrosis.
If the trypsinogen level is elevated and there is one cystic fibrosis gene mutation, there is a one in 20 chance that the child has cystic fibrosis. A single gene mutation is not sufficient for cystic fibrosis, but other cystic fibrosis gene mutations exist. The possibility exists that the child could have a second cystic fibrosis gene mutation not identified through the newborn screening test. A sweat test is needed to determine whether or not the child has cystic fibrosis. Genetic counseling for the parents is also recommended. If a child has one cystic fibrosis gene mutation, then one of the parents is also a carrier of the cystic fibrosis gene mutation. The carrier status of the parents can only be determined by further genetic testing.
If the trypsinogen level is elevated and there are two cystic fibrosis gene mutations, the child is presumed to have cystic fibrosis. A sweat test is needed to confirm the diagnosis. Genetic counseling for the parents is also recommended.